Reanny Medical Devices Management Consulting Co., Ltd.
Focused · dedicated · professional
Head Office
0755-27391220
Guangzhou Company
020-82513196

Focused · dedicated · professional
Head Office
0755-27391220
Guangzhou Company
020-82513196

When discussing the key elements that medical device clinical trial management departments should possess, we have to delve into the complexity and rigor carried by this field. The clinical trials of medical devices, as a key link in verifying product safety and effectiveness, not only directly affect the life and health of patients, but also impact the progress and development of medical technology. Therefore, the construction and operation of an efficient, professional, and compliant clinical trial management department should revolve around a series of core competencies and principles.1、 Professional staffing and team structureFirstly, the clinical trial management department for medical devices should have a highly qualified and specialized team. Team members need to have a strong background in medicine, pharmacy, statistics, ethics, and regulations, and be able to fully understand and accurately execute the requirements of clinical trials. The division of labor within the department should be clearly defined, including but not limited to multiple functional positions such as project management, data management, quality assurance, medical monitoring, ethical review, etc., to ensure that each link of the experimental process is under the responsibility of a dedicated person and can collaborate efficiently.2、 Perfect system and process managementSystem is the cornerstone of management, and the medical device clinical trial management department should establish and continuously improve a series of rules, regulations, and operational procedures to ensure the standardization and traceability of trials. These systems should cover multiple aspects such as trial design, ethical review, subject recruitment and management, data collection and analysis, safety monitoring and reporting, quality control and auditing. At the same time, the department also needs to develop emergency plans to deal with possible emergencies and ensure the safety and rights of the subjects.3、 Strict ethical review mechanismEthical review is an indispensable part of clinical trials. The clinical trial management department of medical devices should establish an independent ethics committee or rely on external professional institutions to conduct strict ethical review of each trial protocol. The review content not only includes the scientificity and rationality of the experiment, but also pays attention to the potential risks and benefits that the experiment may cause to the subjects, ensuring that the experiment is conducted on the premise of respecting the rights and interests of the subjects and ensuring their safety.4、 Efficient data management and analysis capabilitiesData is the core of clinical trials, and its authenticity, completeness, and accuracy directly affect the reliability of the trial results. Therefore, the clinical trial management department of medical devices should establish an advanced data management system to achieve electronic collection, storage, processing, and analysis of data. At the same time, the department also needs to equip a professional data analysis team to use statistical methods to conduct in-depth mining and analysis of experimental data, providing strong evidence for product registration and listing.5、 Strong communication and coordination skillsClinical trials involve multiple parties, including sponsors, research institutions, ethics committees, regulatory agencies, and participants. The clinical trial management department of medical devices needs to have excellent communication and coordination skills, be able to handle relationships among all parties properly, and ensure the smooth progress of trials. This includes but is not limited to cooperation negotiations with the sponsor, coordination and cooperation with research institutions, sufficient notification and communication with subjects, and timely reporting to regulatory agencies.6、 Continuous training and educationWith the continuous advancement of medical technology and the updating of regulations and policies, clinical trial management departments need to maintain a keen perception and learning ability towards new knowledge and technologies. Therefore, the department should regularly organize internal training and external communication activities to enhance the professional competence and comprehensive ability of team members. At the same time, science popularization education should also be carried out externally to enhance public awareness and understanding of clinical trials.7、 Emphasize innovation and scientific researchInnovation is an inexhaustible driving force for the development of clinical trials for medical devices. The clinical trial management department should actively encourage and support team members to participate in scientific research projects and academic exchange activities, and promote the development and application of new technologies and methods. By continuously innovating and practicing, we aim to improve the quality and efficiency of clinical trials, contributing wisdom and strength to the development of the medical device industry.In summary, the medical device clinical trial management department should have professional personnel configuration and team structure, sound system and process management, strict ethical review mechanism, efficient data management and analysis ability, strong communication and coordination ability, continuous training and education, as well as a focus on innovation and scientific research and other aspects of literacy and ability. Only in this way can we ensure the standardized, safe, and effective conduct of clinical trials, contributing to the advancement of medical technology and the health and well-being of patients.
649
Clinical trials of medical devices are a crucial step in ensuring their safety and effectiveness, and ultimately obtaining market access. This process involves multiple stages and complex steps, from the initial planning of the experiment to the submission of the final report, each step is crucial. The following is a detailed explanation of the entire process of clinical trials for medical devices.1、 Research design and scheme formulationThe first step in clinical trials of medical devices is research design and protocol development. The core of this stage lies in clarifying the purpose of the trial, research questions, inclusion and exclusion criteria for subjects, setting of experimental and control groups, clinical endpoint indicators, and sample size calculation. Research design should be scientifically reasonable, feasible, and strictly adhere to ethical principles. The development of experimental plans should be based on guiding principles and product characteristics to ensure a comprehensive evaluation of the performance and safety of medical devices.2、 Ethical review and approvalBefore conducting clinical trials, the research protocol and related documents need to be submitted to the local ethics committee for ethical review. The ethics committee will evaluate the ethical compliance of the trial to ensure that the rights and interests of the subjects are fully protected. This step is an important part of ensuring the legality and ethics of clinical trials. After obtaining approval from the ethics committee, the trial can proceed to the next stage.3、 Recruitment and screening of subjectsAccording to the predetermined inclusion and exclusion criteria, the research team began recruiting suitable subjects to participate in the trial. The recruitment process should follow the principles of fairness and impartiality, ensuring that all potential participants have equal opportunities to participate. Participants need to undergo pre screening, enrollment evaluation, and sign informed consent forms to ensure that they fully understand the purpose, process, potential risks, and benefits of the trial, and voluntarily participate.4、 Experimental implementation and data collectionThe implementation phase of the trial is the core link of clinical trials for medical devices. According to the research protocol and approval from the ethics committee, the research team used and observed medical devices in the experimental and control groups, and collected relevant clinical data and results. Data collection can include various forms such as clinical examinations, laboratory tests, and imaging evaluations to comprehensively reflect the effectiveness and safety of medical devices.5、 Data Management and MonitoringTo ensure the accuracy, completeness, and confidentiality of the data, the research team needs to establish a strict data management system. During the data collection process, data monitoring and quality control are necessary to ensure the rigor of the experimental process and the reliability of the results. This includes measures such as regular data verification, outlier handling, and data backup.6、 Data analysis and result evaluationAfter the experiment, the research team will conduct statistical analysis on the collected data. This includes various methods such as descriptive statistics, survival analysis, and inter group comparisons to evaluate the effectiveness and safety of medical devices. At the same time, the research team also needs to explain and discuss the results, and write experimental reports. The test report should record in detail the purpose, methods, results, and discussion of the test, as well as the evaluation and conclusions of the medical device.7、 Test termination and result reportIf adverse events or unacceptable risks occur during the trial, the research team may need to terminate the trial in advance. After completing the experiment, the research team needs to write an experimental report based on the results, and make the results public and available for publication. This process helps the academic community and regulatory agencies understand the performance and safety of medical devices, providing scientific basis for product launch.8、 Subsequent monitoring and safety assessmentEven if medical devices pass clinical trials and obtain market approval, their safety and efficacy still need to be continuously monitored. The research team and relevant regulatory agencies need to establish a safety monitoring system to monitor adverse events and safety issues during the use of medical devices. Once potential safety hazards are identified, timely measures should be taken to protect patient safety, and product usage guidelines should be adjusted or recalled.9、 Quality Management System and Regulatory ComplianceThe entire process of clinical trials for medical devices must comply with relevant regulations and guidelines. The research team needs to establish an appropriate quality management system to ensure the accuracy, reproducibility, and compliance of the testing process. At the same time, it is necessary to maintain communication and coordination with the ethics review committee and regulatory agencies to ensure the scientificity, compliance, and quality of the experiment.In summary, clinical trials of medical devices are a complex and rigorous process that involves multiple stages and stages. Each step requires strict adherence to relevant regulations and standards to ensure the scientific, compliant, and quality of the experiment. Through this process, the performance and safety of medical devices can be comprehensively evaluated, providing patients with safer and more effective treatment options.
507
In today's rapidly advancing medical technology, medical devices, as an important component of the healthcare system, have a direct impact on the safety, effectiveness, and compliance of patients' lives, health, and medical quality. Therefore, the registration and application process for medical devices is particularly important. It is not only a necessary path for products to enter the market, but also an important link in ensuring public health and safety. This article aims to elaborate on the requirements and instructions for medical device registration application materials, providing a comprehensive guide for relevant enterprises and practitioners. 1、 Overview of Medical Device Registration ApplicationMedical device registration application refers to the process in which an enterprise submits a product registration application to the National Medical Products Administration (NMPA) or the corresponding provincial drug regulatory department in accordance with relevant laws and regulations, and provides a series of documents proving the safety, effectiveness, and controllable quality of the product. This process involves multiple stages, including product classification definition, preparation of registration materials, submission for review, on-site inspection (if applicable), technical evaluation, administrative approval, and issuance of registration certificates.2、 Basic Requirements for Medical Device Registration Application Materials1. Basic product informationProduct name: It should be accurate, clear, comply with naming conventions, and avoid misleading.Model specifications: List all models and specifications in detail, and clarify the differences between each model.Classification code: Determine the management category and classification code of the product according to the "Classification Catalogue of Medical Devices".Production enterprise information: including enterprise name, registered address, production address, contact information, and production license information.2. Technical documentationProduct description: Detailed description of the product's structural composition, working principle, intended use, usage restrictions, etc.Design and development documents: including design input, output, verification, confirmation, and change records, etc., to prove that the product design is reasonable and scientific.Production process: Elaborate on the production process, key control points, raw material sources, and quality control standards.Performance research data: including research data on physical properties, chemical properties, biocompatibility, stability, etc., to demonstrate that product performance meets standard requirements.3. Safety and effectiveness evaluation materialsPreclinical research: including animal experiments, in vitro experiments, etc., to evaluate the safety and preliminary effectiveness of the product.Clinical trial data (if applicable): Detailed records of the design, implementation, results, and conclusions of the clinical trial to demonstrate the safety and effectiveness of the product.Risk assessment report: Identify, analyze, and evaluate risks throughout the product lifecycle, and propose control measures.4. Quality Management System DocumentsQuality Manual: An overview of the structure, responsibilities, procedures, and requirements of the enterprise's quality management system.Program file: specifies the processes, methods, standards, and record requirements for various quality activities.Registration self inspection report (if applicable): The enterprise conducts a comprehensive self inspection of the product in accordance with the registration self inspection requirements and issues a report.5. Other materialsProduct manual and label samples: The content should be accurate, complete, and comply with relevant regulatory requirements.References: List all cited domestic and foreign standards, literature, data, and other materials.Commitment letter: The enterprise makes a commitment to the authenticity, accuracy, completeness, and compliance of the submitted materials.3、 Precautions for preparing registration and application materials1. Compliance with regulations: closely monitor and follow the latest laws, regulations, and guiding principles related to medical device registration to ensure that the information meets the latest requirements.2. Data integrity: Ensure that all necessary information is complete and without omissions, to avoid delays in the review process due to incomplete data.3. Data authenticity: All research data, experimental reports, etc. must be authentic and reliable, and must not be forged or tampered with.4. Clear logic: The organization of data should be well-organized, logically rigorous, and easy for reviewers to understand and evaluate.5. Language standards: Materials should use standardized language to avoid using vague, ambiguous, or undefined professional terms.The registration and application of medical devices is a complex and rigorous task, which not only tests the technical strength and management level of enterprises, but also concerns the health and safety of the public. Therefore, enterprises should attach great importance to registration and application work, strictly prepare materials in accordance with regulatory requirements, and ensure the safety, effectiveness, and compliance of products. At the same time, with the continuous advancement of medical technology and the continuous improvement of regulatory systems, enterprises need to continue to pay attention to industry trends, adjust and improve their registration and application strategies in a timely manner, in order to adapt to market changes and regulatory requirements.
525In the vast field of the medical industry, medical device consulting services play a crucial role. These services not only provide comprehensive support for medical device manufacturers, distributors, medical institutions, and regulatory agencies, but also promote the healthy development of the entire industry. With the advancement of technology and the increasing integration of the global market, the types and scope of medical device consulting services are constantly expanding and deepening. This article will explore the specific types and core values of medical device consulting services from multiple dimensions.1、 Clinical Research Organization (CRO) ServicesClinical research organizations are specialized service organizations that provide clinical trial support for medical device manufacturers. Their core services include clinical trial design, management, case recruitment, data management, and supervision. CRO companies, with rich clinical trial experience and professional teams, can assist manufacturers in efficiently and compliantly completing clinical trials, ensuring product safety and effectiveness, and accelerating the market launch process. In addition, CRO is responsible for organizing and analyzing experimental data, providing solid data support for product registration.2、 Market access and registration consulting servicesMarket access and registration consulting services are indispensable for medical device manufacturers to enter domestic and international markets. This type of service covers research on regulatory standards in multiple countries, research on product regulatory registration strategies, preparation of registration documents, submission and approval follow-up of registration applications, etc. Consulting firms can provide customized solutions to meet the regulatory requirements of different countries and regions, helping manufacturers smoothly obtain registration certificates, business licenses, and production licenses. For example, consulting firms can provide comprehensive registration agency services for international mainstream markets such as SFDA in China, FDA in the United States, and CE in the European Union to ensure product compliance and market launch.3、 Quality management and compliance consulting servicesQuality management is a key link in the production process of medical devices. Quality management and compliance consulting companies are committed to helping manufacturers establish and implement quality management systems that comply with international standards, such as ISO 13485, YY/T 0287, etc. These services include consulting, auditing, training, and improvement suggestions for the quality management system. Through professional training and guidance, consulting firms can enhance manufacturers' quality management capabilities, ensuring that every aspect of product development and production complies with relevant regulations and standards. At the same time, consulting firms can also provide timely and effective response strategies for unexpected events such as adverse events and recalls.4、 Technical evaluation and certification servicesTechnical evaluation and certification services are important means of assessing the performance and safety of medical device products. This type of service is usually provided by professional technical evaluation and certification agencies, including product performance testing, safety verification, EMC (electromagnetic compatibility) testing, biocompatibility assessment, etc. These evaluations not only help manufacturers understand the true performance of their products, but also earn the trust of consumers in both domestic and international markets. Certification services involve the application and approval of certifications from authoritative organizations such as ISO, CE, and FDA, serving as a "passport" for products to enter the international market. Technical evaluation and certification agencies ensure that the quality and safety of medical device products reach the highest international level through strict testing processes and professional evaluation standards.5、 Supply chain management and optimization servicesWith the rapid development of the medical device industry, supply chain management has become increasingly complex. Supply chain management and optimization services help manufacturers improve operational efficiency and reduce costs by integrating supplier resources, optimizing inventory management and logistics processes. This type of service also covers supply chain risk assessment and response, supplier quality audit and continuous improvement, ensuring the stability and reliability of the supply chain. In the context of globalization, supply chain management and optimized services are key for medical device manufacturers to enhance competitiveness and respond quickly to market changes.6、 Innovation and R&D consulting servicesInnovation is a key driving force for the development of the medical device industry. Innovation and R&D consulting services focus on providing manufacturers with cutting-edge technology trend analysis, innovation strategy planning, new product concept development, and other services. By collaborating with research institutions, universities, and industry experts, these services can stimulate manufacturers' innovative thinking and accelerate the development process of new products. At the same time, consulting services also cover support in intellectual property protection, technology transfer, and commercialization, ensuring that innovative achievements can be smoothly transformed into market competitiveness.In summary, medical device consulting services, with their comprehensive and multi-level service model, provide solid support for the healthy development of the medical device industry. In the future, with the continuous advancement of technology and the continuous expansion of the market, the types and scope of medical device consulting services will continue to enrich and deepen, injecting new vitality into the prosperity of the industry.
531MDSAP, The Medical Device Single Audit Program is a global collaborative project initiated and promoted by the International Medical Regulatory Forum (IMDRF), aimed at meeting the regulatory requirements of multiple participating countries and regions for medical device manufacturers through a single audit, simplifying the audit process, reducing enterprise costs, and accelerating product entry into the international market. The MDSAP certification service has a wide and in-depth content, covering the entire process from audit preparation, on-site audit to follow-up tracking and improvement. The following is a detailed analysis of the service.1、 Preparation before review1. Regulatory Compliance AssessmentThe first step of MDSAP certification service is to conduct a comprehensive regulatory compliance assessment of the existing quality management system of the enterprise. This includes comparing the medical device regulatory requirements of MDSAP participating countries (such as the US FDA, Health Canada Canada, MHLW/PMDA Japan, ANVISA Brazil, TGA Australia, etc.) to evaluate the compliance status of the enterprise in quality management system, risk management, product design and development, production control, clinical evaluation, labeling, post market supervision, and other aspects. Through this evaluation, companies can identify their strengths and weaknesses, providing a basis for subsequent improvements.2. Development of audit planBased on the results of regulatory compliance assessment, the MDSAP certification body will work with the enterprise to develop a detailed audit plan. The plan specifies key elements such as audit scope, audit standards, audit schedule, and audit team composition to ensure that the audit work is carried out in an orderly manner. At the same time, the audit plan will also take into account the specific needs of the enterprise, such as key audit areas and special arrangements during the audit process, in order to maximize the efficiency and effectiveness of the enterprise's participation in the audit.2、 On site audit1. Comprehensive audit executionThe core component of MDSAP certification services is on-site auditing. The audit team is composed of experienced auditors from different participating countries or regions. They conduct comprehensive and in-depth audits of the enterprise based on MDSAP audit standards and enterprise quality management system documents through various methods such as document review, on-site inspection, and personnel interviews. The audit covers all key elements of the quality management system, including but not limited to management responsibilities, resource management, product realization, measurement analysis, and improvement.2. Problem discovery and feedbackDuring the audit process, the auditor will record in detail the issues and non conformities found, and communicate and confirm with relevant departments and personnel of the enterprise. For the discovered problems, the auditor will classify them based on their severity and the degree of impact on product quality and safety, and provide specific rectification suggestions. Enterprises need to take these issues and non conformities seriously, and promptly develop and implement effective corrective and preventive measures.3、 Post audit processing and continuous improvement1. Preparation and submission of audit reportAfter the audit is completed, the MDSAP certification body will prepare a detailed audit report, including an overview of the audit process, identified issues and non conformities, and improvement suggestions. This report will be submitted to all regulatory agencies involved in MDSAP as an important basis for enterprises to obtain MDSAP certification. At the same time, the enterprise will also receive a copy for subsequent rectification and continuous improvement.Implementation and verification of corrective measuresEnterprises need to develop a detailed rectification plan based on the rectification suggestions in the audit report and complete the rectification work within the specified time. During the rectification process, enterprises can seek guidance and support from MDSAP certification bodies to ensure the effectiveness and pertinence of rectification measures. After the rectification is completed, the enterprise needs to submit a rectification report and relevant evidence materials to the MDSAP certification body to prove the effective implementation of the rectification measures.3. Continuous improvement and long-term maintenanceMDSAP certification is not a one-time solution, but a continuous improvement process. After obtaining MDSAP certification, enterprises need to continue to strengthen the construction of their quality management system, continuously optimize management processes, and improve product quality and safety. At the same time, enterprises also need to undergo regular supervision and review by MDSAP certification bodies to ensure continuous compliance with MDSAP standards and regulatory requirements of participating countries.4、 The Value and Significance of MDSAP CertificationMDSAP certification services not only help companies simplify audit processes and reduce audit costs, but more importantly, they provide an international stage for companies to showcase the soundness of their quality management systems and product safety. By obtaining MDSAP certification, enterprises can better meet the high standard requirements for medical device quality in the global market, enhance brand awareness and market competitiveness. Meanwhile, MDSAP certification is also one of the important passes for enterprises to enter the international market, laying a solid foundation for expanding overseas business and achieving global development.In short, MDSAP certification services have rich content, rigorous processes, and strict standards, which are important ways for enterprises to improve their quality management level and enhance their market competitiveness. Enterprises should attach great importance to MDSAP certification work, actively prepare, respond seriously, and continuously improve in order to stand out in the fierce international market competition.
620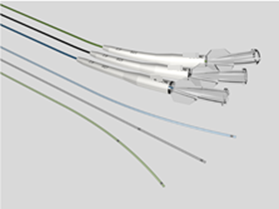
Key points for NMPA registration of microcatheters 1、 Introduction to microcathetersMicrochannels typically consist of a catheter body, radiopaque markers, connectors, and other structures. The tube body is usually thin and soft, and the surface may have a hydrophilic coating. Used for injecting diagnostic reagents (such as contrast agents), therapeutic reagents (such as drug preparations, embolic materials), and appropriate instruments (such as stents, coils) into the vascular system. Microcatheter plays an indispensable and vital role in the field of interventional treatment of coronary atherosclerotic heart disease (CHD), especially in dealing with the challenging chronic occlusive disease (CTO). As the global incidence rate of cardiovascular diseases continues to rise, and patients increasingly pursue the safety and effectiveness of minimally invasive surgery, advanced catheter technology has become a key force to promote medical progress.Microchannels, with their ultra-fine size, excellent flexibility, and precise maneuverability, perfectly meet the needs of complex coronary intervention surgeries. They can easily traverse winding, narrow, and even occluded blood vessel channels, providing doctors with unprecedented treatment pathways and achieving precise navigation and intervention of lesion sites. In the interventional treatment of complex lesions such as CTO, microcatheters not only improve the success rate of surgery, but also significantly reduce surgical risks, alleviate patient pain and recovery time.At the market level, with the continuous innovation of medical technology and the continuous expansion of clinical application scope, the demand for the global microcatheter market is showing a strong growth trend. It is predicted that this market will experience significant expansion starting from $4.3925 million in 2023, and is expected to reach a market size of nearly $8.2469 million by 2032, with a significant compound annual growth rate, reflecting the broad application prospects and huge market potential of microcatheter technology in the field of cardiovascular intervention therapy (data source from the network).In summary, as a key tool in the interventional treatment of coronary heart disease, especially in the treatment of CTO lesions, microcatheters are leading the innovation and development of cardiovascular intervention technology, bringing safer and more effective treatment options for cardiovascular disease patients worldwide.In China, the management category of microcatheters is Class III, with a classification code of 03-13-26 microcatheters. This article provides a brief introduction to the key focus points of domestic registration of microcatheters based on the "Guiding Principles for Registration and Review of Microchannels (No. 4 of 2022)" and combined with the author's previous experience.2、 Reference standards and guiding principlesYY 0285.1-2017 Intravascular catheters - Disposable sterile catheters - Part 1: General requirements;GB/T 16886 series standards;Guiding Principles for Registration and Review of Microchannels (No. 4 of 2022);2、 Requirements for Writing Overview Materials1. Product NameGenerally, "microcatheter" is used as the core word, with structural characteristics, material composition, and usage location as characteristic words, such as "coronary microcatheter".2. Structural compositionNeed to provide axial and cross-sectional views of the microcatheter; If the product is designed with a multi-layer structure, the multi-layer structure should be clearly identifiable on the cross-sectional view.3. Composition materialsFor raw materials that come into direct/indirect contact with the human body, supplier information, manufacturer information, proof of purchase documents, quality control standards, and factory inspection reports are required.For products with coatings, the coating range and method need to be provided; For hydrophilic coatings containing hyaluronic acid, the source of hyaluronic acid should be clearly identified, indicating whether it is extracted from animal tissue or prepared through microbial fermentation.3、 Non clinical data1. Product Technical RequirementsThe performance indicators in the product technical requirements should refer to Table 1 in the attachment of the Guiding Principles for Registration and Review of Microchannels (No. 4 of 2022). If the project is not applicable, the reasons should be explained.For coated products, if the coating material causes abnormal chemical performance results, it is recommended to test uncoated products to confirm whether their chemical properties are acceptable. At the same time, a comprehensive evaluation should be conducted based on the clinical application history and biocompatibility data of the coating material. If the coating can be safely used on the human body and the chemical properties of the uncoated product are normal, this chemical property may not be specified in the product technical requirements. (Tips: Chemical performance testing can be conducted only on the parts in contact with the human body. If the test results are qualified, the evaluation is also acceptable. Therefore, the safety of the product should be analyzed from multiple aspects.). )2. Product performance researchThe performance indicators listed in Appendix Table 1 of the Guiding Principles for Registration and Review of Microchannels (No. 4 of 2022) should provide determination basis and performance research data. If not applicable, reasons should be explained.The performance research data should clearly specify the sample size and the basis for determining the sample size.3. Stability studyThe shelf life verification project includes two aspects: product performance and packaging system performance, among which the product performance should include all the performance specified in the product technical requirements (especially when registering for import)4、 Clinical evaluationMicrocatheters are products listed in the "Catalogue of Medical Devices Exempted from Clinical Evaluation". If they exceed the requirements of the clinical exemption catalogue, they can refer to the "Guidelines for Technical Review of Comparative Clinical Evaluation of Intravascular Catheters of the Same Variety" for clinical evaluation or clinical trials of the same variety.
778Head Office
Headquarters Address:Room 1509, Jingting Building, Dongzhou Community,
Guangming Street, Guangming District, Shenzhen
Phone:+86-0755-27391220
Guangzhou Company
Address:Room 506, No.3-bis, Houwangmiao Street, Shigang Road, Haizhu District, Guangzhou
Phone:+86-020-82513196
Mobile Phone:+8618127257501(WeChat Account)


Phone
0755-27391220
020-82513196
WeChat customer service
Mini Program
reanny@reanny.com