
2017 年5 月5 日欧盟医疗器械法规(MDR)和体外诊断医疗器械法规(IVDR)正式发布,于2017 年5 月25日正式生效,并分别于2020 年5 月26 日(COVID-2019下推迟至2021年5月26日)和2022 年5 月26 日实施。自实施之日起,MDR 和IVDR 将分别取代原欧盟医疗器械指令(MDD)和体外诊断设备指令(IVDD)。

与大部分国家一样,欧盟的监管体系也是呈“金字塔” 式的多层级法规体系: 在医疗领域, 法规层级从高到低依次为法规(Regulation)、指令(Directive)、决议(Decision)等。 IVDR在法规层级上从原先的Directive(指令)上升为Regulation(法规),标志着欧盟当局对医疗设备领域监管的进一步重视,同时也预示着在欧盟各成员国内医疗器械监管的尺度将得到进一步的统一。
1. 定义
IVDR被定义为:是指制造商预期用于体外检查从人体提取的样本,包括捐献的血液及组织,单独使用或组合使用的试剂、试剂产品、校准物品、控制材料、成套工具、仪器、器具、设备、软件或系统,其唯一目的或主要目的是提供以下信息:
·有关生理学或病理学状态;
·有关先天性异常;
·有关健康状况或疾病的易感性;
·确定安全性以及与可能接受治疗者的相容性;
·预测治疗效果或反应;
·明确或监控治疗措施。
2.分类规则的变化以及公告机构介入的增加
2.1产品范围和分类
(第1条,第2条,第22条,第23条,第51条,第52条,附件VIII,IX,X,XVI),基于物质的器械和使用有害物质的器械的严格规则(分类规则) 21和附件I)以及软件和应用程序的新规则 (分类规则11)—在保留分类系统的同时,某些产品的规则收紧和更改,这将导致某些设备被重新分类为更高的类别。此外,某些以前不受法规约束的设备现在已在新法规的范围内。打算通过人体孔口或应用到皮肤上的,由人体吸收或局部分散在人体中的,由多种物质组成的设备或物质组合,将根据不同因素而具有不同的分类。同时,应仔细评估与软件相关的要求,以确定潜在的新分类。
在IVDR法规中,对于体外诊断设备的监管依然是基于分类监管这个大框架,但是分类规则较原先的IVDD却发生了根本性的变化,即在IVDR中,基于产品的风险将所有的体外诊断设备由低到高分为了A、B、C、D四类(分别对应最低级风险到最高级风险),B类到D类产品的技术文档要经过公告机构的评估。
该分类规则来源于全球协调工作组(GHTF),目前加拿大、巴西、澳大利亚等国已使用,是国际范围内认可度较高的分类规则。各类产品举例见表1。

2.2 公告机构的介入
伴随着产品分类规则的调整,各类别产品对应的认证途径自然较原监管体系也有着很大的变化,其中最核心的变化点在于公告机构(Notified Body,NB)介入的增加,涉及产品包括所有的D、C、B类和部分A类。
在整个IVD领域,涉及公告机构介入的产品数量从IVDD监管体系下的10%~20%增加至80%~90%。公告机构介入量的增加,意味着绝大多数的体外诊断设备,在欧盟区的市场准入将要告别原先“自我宣称”的形式,取而代之的将是一个实质性的注册过程。
3.欧盟医疗器械数据库的建立以及监管透明度的增加
在欧盟过去十多年的医疗器械监管中,监管的透明度一直是业内诟病比较多的一点。究其原因主要在于没有一个公开的数据库供各方查询。在IVDR的法规体系下,欧盟主管当局一个很重要的工作就是未来将要推出医疗器械数据库(Eudamed),该数据库涵盖从产品上市前审批到上市后监管中的很多重要信息。见下图。
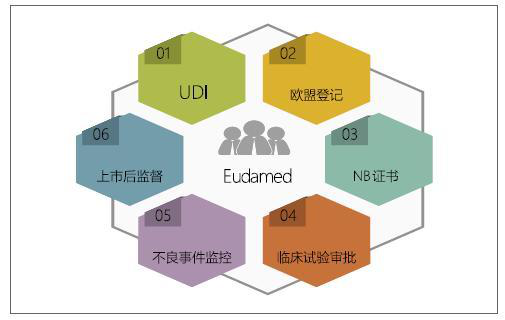
4.唯一设备识别系统(Unique Device Identification System, UDI)的引入
(UDI)/植入卡(第27条,第87条,第18条,第19条) —将引入UDI以在所有医疗设备上提供可追溯性,并将其放置在设备的标签上(而不是运输容器)。UDI将在警惕性报告中引用,并将用于报告严重事件和现场安全纠正措施。此外,所有可植入医疗设备都需要带有警告的植入卡,有关预期设备使用寿命的信息以及必要的后续措施(以及其他要求)。
继美国FDA实施UDI的要求以来,世界范围内纷纷效仿,很多国家相继推出相关草案。欧盟本次出台的IVDR,同样也引入UDI的要求,以增强产品的追溯以及上市后的管理。与FDA的要求一样,IVDR中所提出的UDI同样是由一个固定的产品识别码(Device Identifier,DI)和一个非固定的生产识别码(Production Identifier,PI)组成。生产企业在实施UDI的过程中,需要明确各产品的UDI代码及所包含的信息,并在产品上加贴UDI标贴,同时以电子形式存储UDI相关信息并在Eudamed系统上申报。
5. 分销商和制造商的要求
新法规强调对供应链的格外监督,包括公告机构对关键供应商和分包商的突击审核。此外,新法规规定了进口商、分销商和授权代表的监管作用以及对他们的要求。进口商是指在欧盟范围内建立,将第三国的器械投放在欧盟市场的任何自然人或法人。分销商是指除了供应链的制造商和进口商之外,使得器械在市场可获得的任何自然人或法人。在某些情况下,进口商也可充当分销商。
制造商必须为其产品投保责任保险;但是,进口商有责任验证保险是否充分,否则进口商必须购买额外的保险。除了法定制造商,授权代表必须指定专人负责其组织内部的法规遵从工作。这个专人必须在IVD领域受到过适当的教育以及具有IVD领域的经验。而且,他还必须对技术文档负责,确保符合性声明、性能评估和监测要求及时更新。
此外,新的IVDR对伴随诊断作了特别的规定。其中最主要的一条要求是,由公告机构审核完技术文档后必须向主管当局咨询。我们预计在IVDR正式发表之后,有关伴随诊断的具体指南也将发布。
5.1 经济运营商的角色
(第10、11、13、14、30条)和负责监管合规的人员 (第15条)-经济运营商包括制造商,分销商,进口商,供应商,分包商,组装商和欧盟授权代表,所有这些人承担遵守法规的责任。新的立法条款描述了必须履行的义务,包括“什么”和“如何”,这意味着所有利益相关者的责任大幅度增加。此外,组织内必须有负责法规遵从的人员,并且具有通过经验或资格证明的医疗设备专业知识。
5.2 制造商法规负责人
IVDR法规中,首次提出了法规负责人的要求:要求每个制造商企业内,至少任命一位法规负责人,负责处理与产品相关的监管、合规性相关工作。具体的职责包括产品批放行、起草和维护CE技术文档、完成上市后产品的监控、临床试验相关文件的签署等。
提出法规负责人的要求,标志着新法规IVDR对于执行层面的思考;意味着法规要求在企业内的责任明确、落实到人,从生产企业内部促进着产品的安全性和有效性。
6.法规过渡期
新法规IVDR与IVDD相比变化很大,为避免由于法规切换而造成对现有医疗系统的冲击,实现法规的“软着陆”,IVDR法规从生效(Entry into Force)到实施(Application)期间有5年的过渡期。IVDR法规的生效日为2017年5月25日,实施日为2022年5月26日。在生效日前依照旧法规IVDD颁发的CE证书,在证书有效期内持续有效;在生效日后依照旧法规IVDD颁发的CE证书,在实施日后的2 年失效(若证书有效期晚于该时间点)。
所以,在IVDD法规框架下“自我宣称”类的产品,由于不涉及CE证书,故需要在实施日前完成IVDR法规的切换;少部分在IVDD法规框架下涉及CE证书的产品,需要在证书有效期内完成IVDR法规的切换,但不得晚于实施之后的2 年。
7.警惕性和售后监视
(第84、85、86、87、88、93条) / EUDAMED数据库(第33条)—与警惕性和售后监视相关的新法规影响了国家主管部门和经济运营商,其中包括建立有关医疗设备。该数据库将捕获设备的注册,经认可的指定机构,证书,严重事件,安全和临床绩效报告(SSCP),定期安全更新报告(PSUR),监视活动,临床调查数据,唯一设备标识信息(UDI),以及单一注册信息可关联制造商,授权代表和进口商的信息。
上市后监督需要进入一个持续的评估和改进周期,该周期链接到对风险管理信息的持续审查,以及对临床评估,安全性和性能的公开摘要的定期更新。该新规定要求对上市后监督,报告,安全性更新报告,对于严重事故和趋势报告(分析负面趋势与风险管理的文件)的管理系统的计划。
8.安全性和临床表现摘要
(第32条)—对于植入式器械和III类器械,制造商必须创建安全性和临床表现的摘要。总结必须以对目标用户和患者(如果相关)清晰的方式编写,并应通过EUDAMED提供。
摘要必须至少包括以下内容:
a.制造商名称和单一注册号(SRN)
b.设备名称和UDI
c.说明,先前的变型,区别,附件和其他打算组合使用的产品
d.目的,适应症,禁忌症和目标人群
e.可能的诊断或治疗选择
f.引用适用的任何协调标准和通用规范
g.临床评估报告摘要(见附件四)以及有关上市后临床随访的相关信息
h.建议的个人资料和用户培训
i.有关残留风险,不良影响,警告和注意事项的信息。
9. 临床评估/上市后临床随访
临床研究(第2条,第55条,第61条,附件XIV,附件XV)—关于临床,性能评估和临床研究的强化规则将要求对临床策略和上市后临床随访计划进行全面审查。在许多情况下,对于当前临床要求的MEDDEV遵从性将不足以遵守新规则。需要为现有设备更新临床证据,并且需要令人信服的明确证据的临床评估摘要需要公开获得。对于III类器械和可植入器械,应至少每年更新一次售后临床随访评估报告,安全性和临床表现摘要(上述)。
在第2条,第61条和附件XIV中定义了与临床数据和临床评估有关的要求。除非适用例外情况,否则必须对III类和IIb类可植入设备进行临床研究,并根据第61条提供合理性。旨在去除或施用药物的可植入设备将受到进一步审查,包括评估临床评估,使用说明和售后临床随访计划。对于这些类型的可植入设备,制造商可以在其临床评估之前请求专家小组的咨询。
必须根据第六章(第61-82条)中所述的要求进行临床研究;公告机构将对临床研究数据进行审查,以确保其符合附件XV的规定。
上市后临床随访是一个不断更新临床评估的连续过程,必须作为上市后监督计划的一部分加以处理,在整个设备的预期寿命内查看数据,以帮助制造商确定与设备相关的风险是否仍然可以接受。
10. IVD产品临床试验标准ISO 20916:2019
ISO 14155作为医疗器械临床试验的国际标准被很多国家采用和借鉴,但该标准并不覆盖体外诊断IVD产品。为了更好地指导IVD产品的临床试验,ISO组织TC212于2019年5月发布了《ISO 20916:2019体外诊断医疗器械 – 使用人体样本进行临床性能研究 – 良好研究实践 》标准。
大多数IVD产品的临床性能研究使用的是临床剩余样本和标本库中的样本,对受试者不会产生额外的风险,对于这样的临床性能研究,ISO 20916规定的要求与医疗器械的临床试验要求(在ISO 14155中有规定)有显著的不同;而少数特殊的IVD产品临床性能研究会对受试者产生风险,按ISO 20916的规定,对这类临床性能研究的要求则非常类似于医疗器械的临床试验要求。
此外,新发布的欧盟体外诊断医疗器械法规(IVDR)勘误也明确指明需遵照ISO 20916进行临床性能研究,这预示着ISO 20916将会成为IVDR的协调性标准,以后欧盟CE认证所需的体外诊断IVD产品的临床性能研究(临床试验)需要参照此标准来进行。
综上所述,满足IVDR法规的要求对各制造商有着不言而喻的巨大作用,各生厂商在未来的几年内都需要对此开展法规跟踪及实施工作。
参考文献:李婧,曾哲,刘继广,彭晖,许勤《欧盟新法规IVR解析》,中国医疗器械信息,文章编号:1006-6586(2018)03-0030-02
Copyright © 2017 深圳市瑞恩尼医疗器械管理咨询有限公司 地址: 深圳市光明区光明街道东周社区璟霆大厦1407室 电话:0755-27391220 粤ICP备17083738号

扫一扫,关注我们
